自20世纪80年代发现被称为磷酸肌醇3激酶(PI3K)的脂激酶家族以来,PI3K已被确认为在细胞存活、增殖和分化过程中起重要调节作用。作为受体酪氨酸激酶(RTK)和G蛋白偶联受体(GPCR)的主要下游效应器,PI3K通过生成可活化丝氨酸苏氨酸蛋白激酶(AKT)及其他下游效应器的磷脂,将来自各种生长因子和细胞因子的信号转导至细胞内信使。肿瘤抑制剂PTEN(磷酸酯酶和张力蛋白同源物)是一类重要的PI3K通路负调控子。最近人类肿瘤基因组学研究表明,在多数人类肿瘤中PI3K通路的许多成员都发生了种系突变或体细胞突变。此外,PI3K和该通路中其他激酶易于进行药理干预的事实,使其成为肿瘤治疗研究中最具有吸引力的靶点。; c% M4 d0 e6 X- X/ N1 A; l. Y
e8 Y+ X! b3 {尽管20年前已发现PI3K能与癌基因结合并激活RTK,但是直到20世纪90年代才确定它与人类肿瘤相关。近期的综合性肿瘤基因组分析结果表明,在肿瘤中PI3K通路中许多成员发生突变,表明该通路在肿瘤发生中有重要作用。" g5 ~" Y( a2 V* y# [
' N+ g3 d5 m6 E& D: }: H0 h渥曼青霉素(wortmannin)和LY294002是第一代PI3K抑制剂,这两种抑制剂对PI3K同工酶没有或很少有选择性,因而毒性较大。许多靶向PI3K的化合物已进入临床,其中大部分是PI3K mTOR双重抑制剂。BEZ235是咪唑喹唑啉衍生物,可通过与ATP结合口袋结合抑制多种Ⅰ类PI3K异构体和mTOR激酶活性,目前已进入Ⅰ期临床试验,用于治疗实体瘤。其他进入Ⅰ期临床试验的有BGT226、BKM120、XL765、XL147、GDC0941、GSK1059615和SF1126等。BGT226是作用广泛的PI3K-mTOR抑制剂;BKM120只选择性地抑制Ⅰ类PI3K酶,对mTOR无抑制活性;XL765和XL147也是Ⅰ类PI3K抑制剂,用于治疗实体瘤;GDC0941是PI-103衍生物,可在纳摩浓度抑制所有Ⅰ类PI3K异构体活性;GSK1059615靶向PI3K,用于治疗实体瘤或淋巴瘤;SF1126是PI3K-mTOR抑制剂,用于治疗实体瘤。. C% I# m! I8 x/ p
AKT是RTK-PI3K复合物下游最关键的近节点,也可作为一种有前景的治疗靶标。目前已开发多种AKT抑制剂,包括基于脂类的磷脂酰肌醇类似物、ATP竞争性抑制剂和变构抑制剂。其中,最有临床前景的是哌立福新(perifosine),可通过靶向AKT的普列克底物蛋白同源物结构域,阻止AKT与PtdIns(3,4,5)P3结合发生膜转位。目前该药进入临床试验阶段,用于治疗各种癌症。多数ATP竞争性小分子AKT抑制剂非选择性地靶向3种AKT异构体。GSK690693是其中之一,不仅可在纳摩浓度下靶向3种AKT异构体,还可抑制来自AGC激酶家族其他激酶活性。通过化合物库筛选及应用迭代虚拟合成,已发现大量变构AKT抑制剂,这类抑制剂有一定的选择性。AKTi-1/2是AKT1和AKT2的双重变构抑制剂,在肿瘤异体移植模型中表现出抗肿瘤活性,AKTI-1/2类似物MK2206用于治疗局部性或转移性实体瘤已进入Ⅰ期临床。XL418是可双重抑制AKT和S6K的小分子化合物,已进入Ⅰ期临床用于治疗实体瘤。VQD-002是水溶性三环核苷酸,用于治疗实体瘤和血液恶性肿瘤,现处于Ⅰ期临床。
9 [4 j ~: I% O. @( ]尽管最近才发现mTOR是PI3K通路成员之一,但它是该通路中第一个用于临床靶向治疗的节点。西罗莫司(雷帕霉素)是一个典型的mTOR抑制剂,是来自细菌的天然产物,最初用于抗真菌治疗,后来发现它具有免疫抑制作用和抗肿瘤活性西罗莫司与其细胞内受体FK506结合蛋白12(FKBP12)关联后直接与mTORC1结合,抑制mTOR介导的下游底物S6K和4EBP1磷酸化。西罗莫司类似物如CC1-779和RAD001已被开发为抗肿瘤药物。ATP竞争性mTOR抑制剂torkinibs和torin1同时抑制mTORC1和mTORC2,可更有效阻断PI3K通路激活,与西罗莫司相比,抑制细胞生长和增殖活性更好。此外ATP竞争性抑制剂OSI-027和AZD805也已进入临床试验,用于治疗实体瘤和淋巴瘤。在PI3K通路中有多个抑制HSP90的化合物如格尔德霉素(geldanamycin)及其类似物,至少部分是通过抑制PI3K通路来达到治疗效果。
+ h2 Z; F+ R- _( F: t2 CPI3K-ERK信号在胰岛素诱导的血管平滑肌细胞增殖中的作用:
, A2 H$ b% V; Y, ^血管平滑肌细胞增殖是动脉硬化和高血压发病机制中的重要环节。近年来研究发现糖尿病伴动脉硬化患者血管内膜、中层VSMCs有胰岛素前体C肽储积,并可刺激单核细胞、CD4+T趋化。胰岛素可明显增加血管紧张素Ⅱ等生长因子对平滑肌细胞的促增殖作用,其介导血管损伤的作用可能通过ERK5信号通路发挥作用。Walcher等发现,胰岛素受体底物Src激酶、PI-3K亦参与C肽介导的促VSMCs增殖,其中Src激酶为PI-3K的上游信号,给予Src-kinase特异阻断剂PP2或转染Src siR-NA可使胰岛素促VSMCs增殖消失,实验中同样发现胰岛素能VSMCs增殖,呈现剂量依赖性,其中10-5mol/L胰岛素促VSMCs增殖幅度上升152%。给予10-6mol/L LY294002抑制PI-3K后,胰岛素促VSMCs的增殖作用下降48.8%,给予10-6mol/L PD98059胰岛素促VSMCs的增殖作用下降43.6%,提示PI-3K、MAPK信号均部分参与了胰岛素促VSMCs的增殖。据报道,胰岛素前体C肽刺激肾小管细胞Na+-K+-ATPase和Swiss 3T3成纤维细胞MAPK需要PKC和PI3K信号分子激活,C肽诱导单核细胞和CD4+T细胞趋化亦需要激活PI3K,Walcher等研究发现,阻断PI3K信号可完全阻断C肽诱导的ERK磷酸化, ERK1/2为PI3K的下游信号分子,实验中发现PI-3K、MAPK仅部分参与胰岛素促VSMCs的增殖,进一步利用Western blotting方法,发现胰岛素刺激SD大鼠VSMCs后,磷酸化ERK1/2蛋白表达增高234% (P<0.05vs对照),阻断PI-3K信号后,胰岛素刺激磷酸化ERK1/2蛋白表达部分被阻断,ERK活性测定结果与Western blotting一致,胰岛素刺激可引起ERK活性增加278% (P<0.05vs对照),阻断PI-3K信号,胰岛素刺激的ERK的活性亦明显降低,提示胰岛素促VSMCs增殖信号通路部分经由PI-3K-ERK1/2通路。5 ` |' I9 b- d. V
PI3K/Akt途径在朊蛋白介导胃癌耐药中的作用研究:
X. R2 g' ?% f% S* U朊蛋白(prion protein, PrP)由美国科学家Prusiner发现并命名,该蛋白存在两种空间构象:细胞型PrPc(cellularprion protein)和致病型PrPsc(scrapie associated prion protein)。研究人员在研究胃癌多药耐药的分子机制中,通过抑制消减杂交技术发现PrPc在胃癌多药耐药细胞系SGC7901/ADR中的表达明显高于其亲本药敏细胞系SGC7901。进一步通过正反义核酸及小干扰RNA技术证实,增强PrPc的表达可以使胃癌细胞的耐药性增加,而抑制其表达,则可逆转耐药胃癌细胞的耐药性,表明PrPc与胃癌耐药表型密切相关。肿瘤细胞产生耐药的机制是十分复杂的,凋亡抵抗是重要机理之一。PI3K/Akt信号途径的激活则又是细胞产生凋亡抵抗并获得生存的关键环节。
/ \1 E* m& s0 y7 C' f1 ]研究人员采用LY294002抑制PI3K/Akt活性,观察其对PrPc介导的胃癌耐药表型的影响,通过细胞水平的耐药表型实验分析转染细胞耐药指数,发现PrPc高表达胃癌细胞对化疗药物敏感性及细胞内阿霉素的蓄积和潴留量均较转空载体对照细胞明显下降,表明转PrPc胃癌细胞耐药性更强,这与我们先前的研究结果是一致的,进一步证实了该分子与胃癌耐药的关系。LY294002可竞争PI3K的ATP结合位点,从而特异的抑制PI3K活性。研究发现当通过LY294002抑制PI3K/Akt途径活性后, PrPc高表达胃癌细胞对对化疗药物的敏感性随着LY294002浓度的增加均逐渐增强,其细胞内阿霉素的蓄积和潴留量逐渐增多,但当LY294002到达一定浓度时, PrPc高表达胃癌细胞的耐药指数接近空载体对照细胞。这就表明,当抑制PI3K/Akt途径活性后, PrPc介导的胃癌耐药特性可以得到逆转,从而证实了PrPc介导的胃癌耐药与PI3K/Akt信号途径活性密切相关。最近有文献报道PrPc与非受体酪氨酸激酶Src共定位于脂筏结构域并激活Src家族激酶,而Src家族激酶可激活下游Ras信号系统并进而激活PI3K/AKT途径。0 a$ H, G5 \$ h3 x: \4 k& r
PI3K通路为肿瘤治疗提供了很大机遇,也面临着巨大挑战。药物研发关键是发现新的治疗靶标替代PI3K抑制剂或在亚毒性剂量时加强PI3K抑制剂疗效。目前仍需要大量研究来确定既能有效阻止肿瘤生长又可使药物副作用降到最小的药物剂量。靶向通路中两种激酶、联合用药及靶向血管生成是未来这类抑制剂的发展方向。 |
 汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
汇总4月最新:临床试验招募| 小细胞
"化疗±免疫后进展了,还能怎么办?"——这是许多小细胞肺癌患者和家属的困境。
临
 新人求助
我爸爸身体不适 26 年 4 月 10 号去医院检查,初步结果是肺肿可能是肺癌,因年纪大 7
新人求助
我爸爸身体不适 26 年 4 月 10 号去医院检查,初步结果是肺肿可能是肺癌,因年纪大 7
 母亲肺腺癌晚期骨转移,EGFR21突变,
母亲1970年出生,56周岁,身高163cm,体重98kg(196斤)。2025年9月23日肺炎住院,胸
母亲肺腺癌晚期骨转移,EGFR21突变,
母亲1970年出生,56周岁,身高163cm,体重98kg(196斤)。2025年9月23日肺炎住院,胸
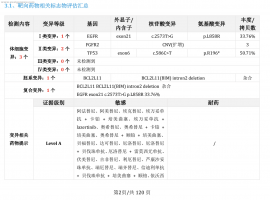 肺癌术后五年,脑膜转移治疗三年,伏
2025年8月14日更新: 终于输液港血培养结果为阴性了,14天美平消炎出院了,昨天开始双
肺癌术后五年,脑膜转移治疗三年,伏
2025年8月14日更新: 终于输液港血培养结果为阴性了,14天美平消炎出院了,昨天开始双
 从6个月到1400多天:一位晚期肺癌患
2026年全国肿瘤防治宣传周(4月15日-21日)即将到来,今年中国抗癌协会的主题是“肿瘤
从6个月到1400多天:一位晚期肺癌患
2026年全国肿瘤防治宣传周(4月15日-21日)即将到来,今年中国抗癌协会的主题是“肿瘤






 显身卡
显身卡






